文章来源于互联网:Nature子刊,字节跳动开发MD模拟预测框架,助力锂电池液体电解质研究

编辑 | 萝卜皮
尽管机器学习力场 (MLFF) 在固体和小分子中得到广泛应用,但在应用 MLFF 模拟液体电解质方面仍存在明显差距——液体电解质是当前商用锂离子电池的关键组成部分。
在这里,字节跳动团队的研究人员提出了 ByteDance AI Molecular Simulation Booster (BAMBOO),这是一种用于分子动力学(MD)模拟的预测框架,并展示了其在锂电池液体电解质环境中的预测能力。
研究人员设计了一个受物理启发的图等变 Transformer 架构(GET)作为 BAMBOO 的主干,以便从量子力学模拟中学习;同时,他们引入了一种集成知识蒸馏方法并将其应用于 MLFF,以减少分子动力学模拟观测值的波动。
他们还提出了一种密度对齐算法,从而使 BAMBOO 与实验测量值对齐。BAMBOO 在预测各种溶剂和盐组合中的关键电解质特性(例如密度、粘度和离子电导率)方面表现出了最先进的准确性。
当前版本的模型针对 15 种化学物质进行了训练,与实验相比,各种成分的平均密度误差为 0.01 g·cm^-3。
该研究以「A predictive machine learning force-field framework for liquid electrolyte development」为题,于 2025 年 4 月 1 日发布在《Nature Machine Intelligence》。

液态电解质是目前大多数商用锂离子电池中不可或缺的组成部分。现有的商用电解质多为碳酸盐基,通常由五种以上的成分组成以满足不同的需求。通过实验探索分子相互作用以合理设计电解质成本高昂、耗时长,并且严重依赖化学家的直觉和经验。
这些限制对从实验室的概念验证实验过渡到市场产品带来了挑战,特别是由于优化多组分液态电解质性能的复杂性呈指数级增长。
当前,比较有潜力的解决方案是用机器学习力场(MLFFs)对液态电解质进行模拟,不过该方法也面临着双重挑战:
一方面需平衡量子力学精度与经典力场效率,通过图神经网络与物理驱动模型融合提升通用性,但离子-溶剂复杂结构(如 SSIP/CIP/AGG 共存)导致跨体系预测困难;
另一方面,现有研究虽验证了特定体系可行性,却缺乏普适性证据,且 MLFFs 普遍存在模拟崩溃、结果波动及过度依赖量子数据而偏离实验的问题,实验数据驱动的参数优化因计算成本与过拟合风险尚未有效实现。
BAMBOO
在最新的工作中,字节跳动的研究人员介绍了 BAMBOO(ByteDance AI Molecular Simulation Booster)工作流程,该工作流程专门用于构建用于有机液体 MD 模拟的 MLFF,特别是液体电解质。

图示: BAMBOO 概述。(来源:论文)
首先,研究人员设计了一个 GET 架构,利用 DFT 计算的知识,将半局部、静电和色散相互作用分开;具体来说,他们将液体电解质中的局部原子环境采样为气相团簇,然后使用 DFT 计算它们的能量、原子力和电荷。
其次,他们在 MLFF 上引入了集成知识蒸馏,抑制了基于 MLFF 的 MD 模拟结果的波动。同时研究人员提出了一种基于物理的对齐方法来协调模拟密度与实验数据,从而建立宏观和微观尺度之间的联系。
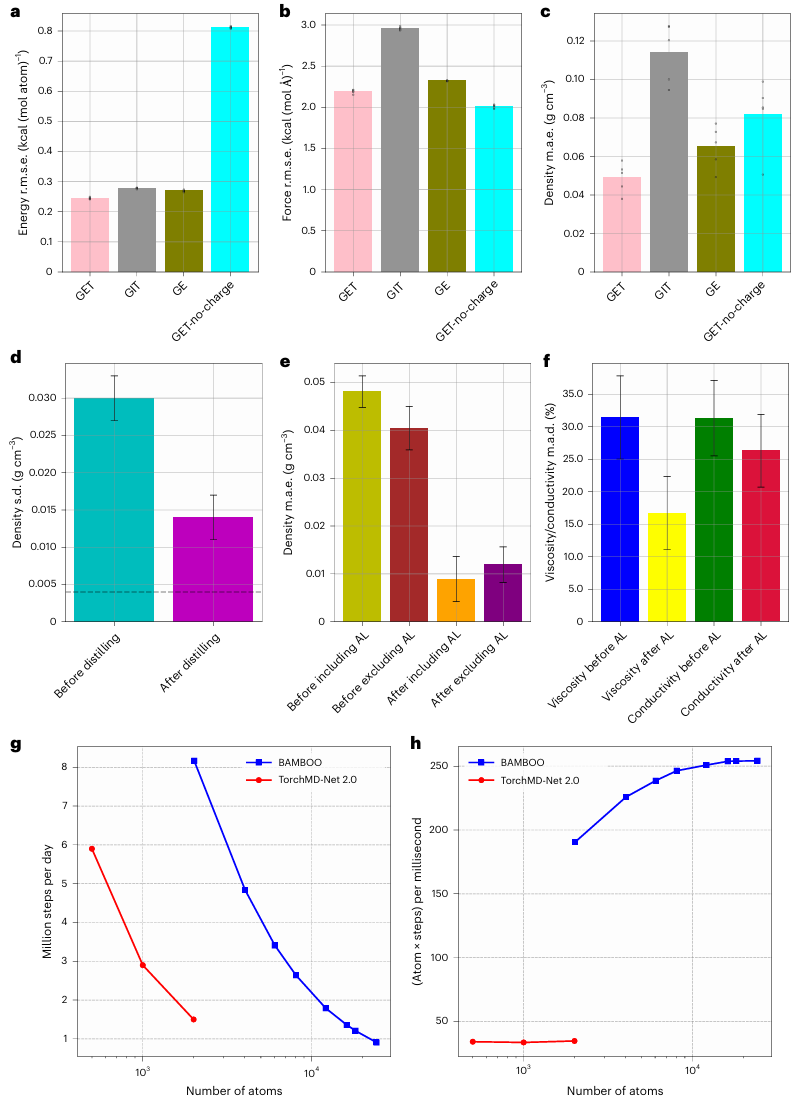
图示:GET 层的影响、集成知识提炼和密度对齐。(来源:论文)
为了保证 BAMBOO 的广泛适用性,该团队在 DFT 数据集中包含了各种分子和盐。值得注意的是,他们的重点涵盖了锂离子电池中使用的液体电解质中常见的成分,例如环状碳酸酯、线性碳酸酯以及 Li+ 阳离子和 PF6-、FSI- 和 TFSI- 阴离子。
此外,研究人员结合了两种常用的有机液体乙醇 (EO) 和丙酮 (ACT),以及工程流体 Novec7000,从而确保训练模型的通用性。
评估结果
研究人员对 BAMBOO 在溶剂和液体电解质上的性能进行了全面评估。模拟结果表明,单个 BAMBOO 模型可以高精度地预测各种化学物质的密度、粘度和离子电导率。
当前的 BAMBOO 模型能够模拟多达 15 种不同成分的物种。这种稳健性凸显了 BAMBOO 在促进实际液体电解质设计和优化方面的价值,这些电解质可能包含多达 10 种成分。
除了预测本体特性的最先进的准确性之外,研究人员还展示了溶剂化结构和原子部分电荷与电解质成分之间关系的量化,为溶剂化工程提供了经典力场或 DFT 无法得出的解析。
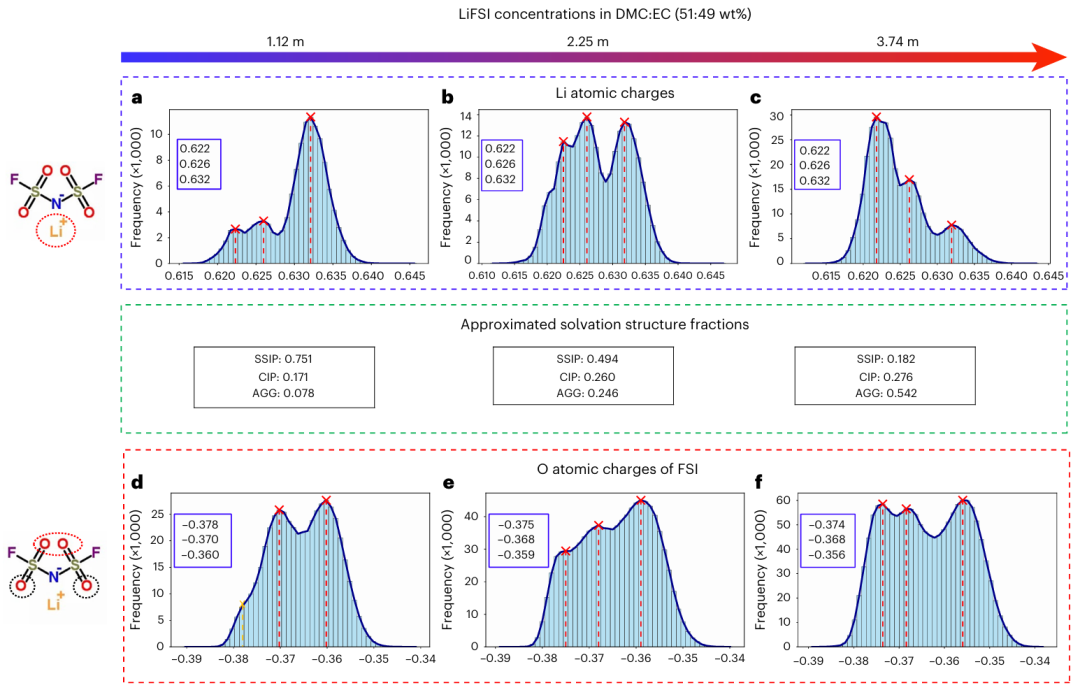
图示:使用 BAMBOO 模拟三种 LiFSI 电解质的原子电荷分布和溶剂化结构分数。(来源:论文)
总而言之,BAMBOO 在预测各种液体电解质的密度、粘度和离子电导率方面达到了最先进的精度。它的高预测能力使其成为由分子结构工程驱动的电解质设计的宝贵工具。
与目前的经典(可极化)力场相比,由于 BAMBOO 是一种没有明确限制函数形式的机器学习力场,因此它有可能扩展到模拟体积特性以外的其他领域,例如模拟液体中的化学反应。
研究人员设想这项工作为开发能够准确模拟大多数有机液体的特性和行为的「通用机器学习力场」奠定了基础。
论文链接:https://www.nature.com/articles/s42256-025-01009-7
文章来源于互联网:Nature子刊,字节跳动开发MD模拟预测框架,助力锂电池液体电解质研究
